
An online presentation with Pipeline.ai about how Google built AlphaFold to produce an end-to-end protein modeling pipeline.
Video • Slides
Introduction (16:45)
Google announced AlphaFold right at the end of last year. They entered it into this online protein prediction contest called CASP. They were able to significantly advance the state of the art in the field. And so I started reading up on the whole thing and it’s a very interesting subject. I started reading more and more, and I thought I would put together this presentation to get all of my thoughts out of my head, and in this way, maybe it can give you an entry point into this subject. There’s a couple of interesting blog posts, one is by this guy named Mohammed AlQuraishi, it’s a very interesting blog post and it’s where I did a lot of my initial deep dive based off of that. I’ll link that here at the end as well. But you definitely should read that.
Obligatory XKCD comic here. First, let me start off with a couple of caveats. One — my background is more on the computers and computer science end of things, with a little bit of the neural networks. I’m not a biologist, so take everything I say here with a very big grain of salt because I’m just abstracting things here. The second thing I’ll add is that Google has not announced a paper for this whole project per se, so the only thing that they’ve actually done for the AlphaFold so far is basically this two-page abstract that they had to submit in order to get into the CASP competition. So basically everything I’m going to be talking about today is going to be me giving educated guesses on top of that two-page abstract. So I’m making good guesses here, but this is very much just a hypothesis. The flip-side of that though is that while I was in grad-school, I took an introductory course to protein modeling as an overview of the field at the University of Missouri, under professor Jack Cheng there. He’s one of the people that makes entries to CASP every couple of years. I think I have a better background on this subject than the average layperson.
Overview of Protein Modeling & AlphaFold (19:44)
So high level — we’ll do a quick review of protein modeling, we’ll discuss the CASP competition and the AlphaFold results, we’ll look at a high-level overview of how the AlphaFold prediction pipeline works — basically, it’s a combination of a couple of different tricks. They use three different neural networks. The first level is using something called DRAW which is a VAE (variable auto-encoder) that they’re using to generate fragments based on the torsion backbone. From there, they combine the fragments together using traditional simulated annealing. From there, they can score the proteins using a combination of two different techniques. One is based around coevolutionary residues, and the other one is scoring networks, a 3D based approach. Finally, they take this combined protein model and they apply some energy models and improvements based on that in order to get their final protein. So we’ll go through all of these steps in this order and I’ll do a demo at the end of where you can go from here.
A Deep Dive into Protein Modeling (21:05)
So protein modeling… DNA makes up all proteins. DNA sequencing has gotten very cheap over the last few decades, and so there are tons and tons of DNA that has been sequenced, various proteins have been sequenced. On the flip-side, we don’t have a ton of great models of how things work. So in an ideal world, we can somehow take our DNA sequence out of our sequencing machine, put it into some sort of magical black box, and then we would have a model of the actual protein on the other end. The most practical and the usual real-world example of this stuff is just predicting drug interactions. Say you have a perfect model of your protein, and then imagine a chemist can look at that and say, “Ah! I can fit in my drug here to say bind this particular receptor,” or something like that. The other thing that’s interesting is that a lot of people are trying to build new proteins now. This is a new and exciting area — basically, you can generate a sequence of DNA and send it to a DNA sequence lab, and they can return you a test tube with that protein. So if you could somehow do this process computationally, then you can save yourself doing the real world round trip, so to speak. There’s a lot of experimental processes for doing protein modeling, but they’re all extremely expensive. We’re talking tens of thousands if not hundreds of thousands of dollars in order to do so. So in an ideal world, we would just magically take our input DNA sequence and add it to our computer, it would produce some sort of model, and then we could do science cheaply. Then we could make money off the whole thing.
Two Schools of Thought on Protein Modeling (23:16)
 Ab Initio / Homology
Ab Initio / Homology
First, we’ll do a very quick overview of protein modeling. Very broadly, there are two basic schools of thought on how to do protein modeling. The first one we’ll say is the physics-based world. We can model atoms and whatnot now extremely well using modern physics. In theory, proteins are just collections of atoms, so we should be able to model proteins as an extent to this. This is a whole area of modeling; it’s deriving things from first principles, that’s the ab initio. Then the flip side is homology or template modeling. Basically, we take real-world data and we try to make predictions from that. So we might take one protein and say that it looks half like one protein and half like another protein, and so if we could just literally take the two models and chop them in half and glue them together, we would actually have a reasonable prediction for what our new protein would look like. In practice, most things in the real world end up in some sort of combination of these two things. Ab initio works well in small scales, but once proteins get large, it starts to become extremely computationally intractable, we’ll say. Homology — if you have a protein that looks very similar to something that’s been found before, you can do stuff with that — but oftentimes, the further and further away you get from traditional approaches or existing stuff, the more and more you’re just starting to guess. The other reason I picked this slide here is because it’s sort of demonstrating end to end protein modeling, which is a subject we’ll come back to at the end.
The CASP Competition (25:25)
CASP is a competition where they test people’s ability to model proteins. It happens every two years, and there are groups from all over the world that participate in this. The basic idea is that they pick some proteins that haven’t been modeled before or haven’t been sequenced, they give it to the groups, and a third party goes off and actually does the physical work to build a physical model. They come back together and try to see how close the predictions matched reality. There’s been slow and steady progress in this field, it’s been going on for a long time. If you look at this graph over here, this is from Mohammed’s blog post. You’ll see that the dotted line is the expected improvement and with his conceptual model, AlphaFold has roughly doubled what the normal progress in this field would be. So this graph gives you a picture of what happened there. In an ideal world, we would be up here at 100% accuracy, so there’s still a really long way to go. But it’s cool that this amount of progress has been made in this short amount of time. So now we’ll try to break down how they did it.
Breakdown of AlphaFold (26:06)
The AlphaFold is an end to end pipeline. So it starts off with a sequence, and then it outputs at the very end a full blown protein model. It’s composed of these different elements. The first one is a neural network that’s based around what’s called the DRAW model, we’ll look at that in a second. But this generates fragments — we’ll say with 3 and 9 residue protein sequences. From there, these fragments are combined together using simulated annealing, which is a traditional approach in this field. From there, AlphaFold uses two different neural networks. One network is based around this concept of inter-residue distances, we’ll look at that in a second. The second one is a scoring network; it attempts to quantify how much like a real protein the protein looks. Then a combination of these two neural networks is used as the score to produce a candidate protein. From there, they add relaxation, which is a common approach in this field. Then finally, they have one more neural network to do a final scoring of the protein, and then output it to produce the end results.
The DRAW Model (28:50)
The DRAW model — I thought this paper was really interesting. This is from the original paper, it’s from a few years back. But basically they combine an attention model with convolutional neural networks, and so the network can be trained to generate realistic looking input coming from raw noise. They use an attention model on top of a variable autoencoder. They feed it input data, and then the network can refine any new input into one of the outputs that it’s seen before. They took this approach, which was applied to 2D images in its original paper, but they applied it in a completely different domain of 1D residues. So they can take the protein fragments from the real world and model them as a collection of backbone and torsion angles. This is a well-known approach in the field, but they can then feed this VAE large amounts of real-world protein data, and it can then start to generate realistic looking fragments based on whatever it needs, so to speak. So then this allows them to generate a lot of candidate fragments for whatever combination of input residues they need. From there, they can take these fragments and combine them together using simulated annealing.
This is a well-understood, traditional approach in this field, but we can take these fragments and then try to minimize an energy function across the collection of inputs. From that, we end up with a candidate full-blown protein string. This is what’s usually called protein folding, but this is just a good way of getting from fragments to something that might actually start to look like a real-world protein fragment.
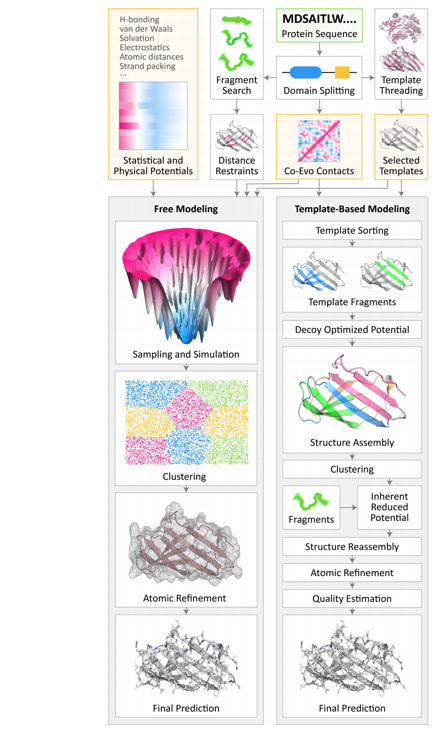
On top of this, the AlphaFold engine adds a couple of interesting tricks. The first one is this use of coevolutionary statistics. Basically, whenever proteins fold, various regions have a higher probability to bind together or come into contact. If we look at this first one for (a), this is what a real-world protein would look like and model for. But then we can pull apart the protein and produce this contact map. Loosely, this is just a large diagonal. Then all these dark areas are regions that are associated together. You can see there are patterns, there are areas that like to bind together. In the real world, these correspond with low energy states for the proteins, or by extension, where whenever the protein binds together, places where it’s most likely to go. This contact map is an image, and we can start to model it using convolutional neural networks. As a random aside, there was a really interesting paper I found in this field — essentially they took contact maps for all these different proteins and then they applied a GAN to it, and then the GAN could reproduce or generate synthetic protein contact maps, and by extension, something that you can model to an actual protein just based on whatever input it was given.
 Deep Contact
Deep Contact
This is deep contact. This is a paper from a couple of years ago, 2018 or so. It builds up a convolutional network to work with these coevolutionary contact maps. At the top is how the actual convolutional neural network works. They simply input this contact map as a 1D map, apply just a very basic 1D CNN transforms to it, some paths, they concatenate things together and do some 2D convolution tricks, and then finally they can output a prediction. I like this paper just because I’ve done a lot of CNN’s. I felt like I could wrap my head around this.
[Brett: I got these two papers swapped. Deep Contact is from 2018 and the De Novo paper is from 2016. I removed some incorrect text from the transcript.] This is a different paper with the same approach here. So this is a much prettier graph of how the pipeline works: loosely, we take an input alphabet of sequences, run them through a residual network, generate this sort of thing, and then output our new predicted contact map.
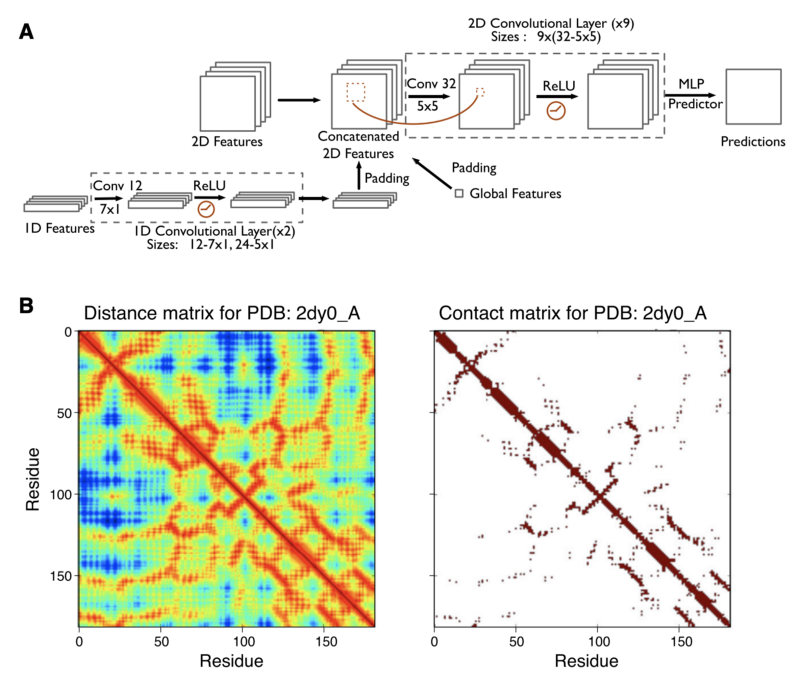
 Scoring Network
Scoring Network
The second neural network that’s done is a scoring network. This is more of me guessing, but it’s my belief that they’re using some sort of 3D convolution based off of real-world data. They can take lots of real-world DNA sequences and run them through 3D transforms in order to produce a score of how much a candidate protein looks like a real-world protein, or like real-world proteins we’ve seen before. So I believe they’re simply using some version of this approach, and they’re running a lot of data samples through it. We’ll come back to that one here in a bit, but I think this 3D CNN approach is probably part of their tricks. So by combining the contact maps with the 3D scoring here, basically they’re able to generate a protein using simulated annealing underneath it that should be reasonably well-built together.
The problem then is oftentimes these protein models are over bound. As a result of doing the math — the math is often optimized towards getting to a minimum energy state, but in the real world with the protein models, that’s not usually the case. Oftentimes, as a result of running these first few steps, you end up with a protein model that’s all lobbed together. So this is a picture from a Zhang lab paper from a few years ago. The basic idea is to take the protein model and to loosely relax it, or just to add a little bit of an energy system and let it bounce around a little bit so that it gets to a state where it’s not overfitting to the problem, so to speak. I think that’s the best way to describe it. Sometimes you’ll see this referred to as refinement in the paper, but these terms are basically interchangeable. This is a well-known traditional thing, but it’s an important part of the protein pipeline.
Then finally they have one more network to do final scoring of the thing and produce the output results. For this, I don’t really have a clear picture of what’s going on conceptually, so I’m extrapolating here. The Mohammed AlQuraishi blog post mentioned this other energy network right here, this NEMO. This is from a different group, but there’s a high probability of the AlphaFold people doing something similar to what NEMO is doing for the final energy prediction. But basically, they’re using a combination of these other steps that I’ve gone through before, and then some real-world physics to try and come up with some sort of number that is an approximate prediction for the value of the network. This NEMO one is interesting to look at. We’ll get into more as to why in a second. But loosely, they’re trying to build these protein simulation models and go from the first step to the end step. So instead of doing a step and feeding it into a pipeline, we can model things from end to end. Then, in theory, the holy grail would be that you can reduce all of this computational complexity because you’re only building one gigantic model. I think this goal is a ways off yet, but I think the AlphaFold results are an important step towards trying to unify this process.
Recap of AlphaFold (39:32)
Very broadly, we’ve looked at how AlphaFold is put together. It starts off with some fragments, then we combine the fragments using simulated annealing. We score the results based on both coevolutionary statistics, which is based off of real-world protein data, with a 3D modeling function which is based off of real-world samples. Then finally, we relax the model and run it through another scoring network in order to get our final results.
Whenever I do these presentations, I like to do a demo. What I thought I would demo here is a 3D CNN. I think a lot of people are familiar with how 2D CNN’s work and conceptually 3D CNN’s are just adding another dimension to the process. But historically, they’ve been extremely computationally expensive, so people have stayed away from them. You see them here and there in the medical world, but often the problem there is that the medical world doesn’t really have enough data in order to really use these things in the real world.
 3D CNN
3D CNN
But anyway, we’ll run through this demo here. It’s a 3D CNN applied to the MNIST data set. This guy took the MNIST data and generated 3D voxels based off of the things… I’m just running it here locally on my device. We’ll let it run here in the background, it takes a couple of seconds. Here’s your traditional 2D MNIST data set, and he generated these 3D voxels. Then we take a two dimensional CNN approach, and then layer it in on top to recognize the data. This model does pretty well, but it’s cheating — we’re using synthetic data, it’s way cleaner data than anything you would probably encounter in the real world. Of course, it fails on a demo… We’ll load it back up here in a second. This other slide on the far side is Voxnet. These 3D CNN’s are used a lot in LiDAR applications right now. So this is kind of an interesting real-world area where this stuff is starting to be used.
Recap of Protein Modeling (43:30)
Very broadly — we’ve taken a quick look at protein modeling in general, and how it works. We’ve done a high-level overview of how the AlphaFold builds an end to end protein modeling pipeline. What’s interesting about this approach is that I don’t think it’s as revolutionary as perhaps some people in the press made it out to be, but on the flip-side, they came in and built on top of a lot of existing stuff that had already been done before, and then found some ways to significantly advance some things. What I thought was really interesting about the AlphaFold approach is that basically, they’re building an end to end pipeline. A lot of the stuff in protein modeling has historically been, “Use one tool to generate one result, then send it to another tool which generates another result, which you send to another tool which generates another result.” So you’re doing this alchemy of combining things tougher. This creates a lot of problems. One of the big ones is that simply that not all the tools play together, so you’re just sitting there troubleshooting weird stuff, like… One tool deals with pi in radians, and another tool deals with pi in degrees, and you’re sitting there dealing with issues as a result of that. So what’s cool to me about this AlphaFold is that they take something in at one spot and it comes out the other, and every part in between is basically completely deterministic. There’s no magic involved. The second interesting thing about this approach is that traditionally whenever people approach these problems, say you’re modeling a particular protein — as a result, most of your data then becomes related proteins in the field. So I think that this AlphaFold approach is interesting because it’s basically a full-blown big data style approach, we’ll say. They can basically take the entire PDB database and run it through the system, and then the more proteins it’s seen, the better the final end results will be. That’s interesting to me because as this field gets more and more data, as the data complexity continues to expand in the next few years, their models are going to improve as more and more things get sequenced and as more inputs come into the field.
Conclusion (46:27)
So broadly, from my conclusion, I would say that this is a small field that needs more eyeballs on it. Things are starting to finally approach the point where lay people can come in and mess with the tools, so if they can provoke more interest in this field and get more people playing with this stuff, I think that ultimately is what’s going to significantly move the needle down the road.
We’ll see if my demo had any luck… We’ll give it one more try. My demo got about 99% accuracy on that dataset, but it’s dealing with synthetic data, so it’s kind of cheating. But anyway, thanks for listening to me. If you’re interested more in this subject, I would highly recommend you read this blog post by Mohammed. He has this YouTube video that he published last month that you should check out. Also, there’s this YouTube video here from last fall explaining how the NEMO system models proteins, which I thought was interesting as well. Thank you for your time.
Questions and Answers (48:00)
Sweet, man. That was good stuff. Is it safe to say that most of Deep Minds models now come out in TensorFlow form? So they’re reproducible, and pretty well documented?
I would say yeah; they’re definitely doing things internally (with TF). They’re definitely extremely interested in commercializing all this stuff. Historically, they would publish a bunch of papers, but I think they’re probably much more interested in seeing how they can make some money off of this. But I’d say they’ve done a lot of work on this, so they’re entitled to it.